
In order to assimilate large data sets generated by high throughput experiments, scientists at NCCS have developed a novel algorithm solving an NP (non-deterministic polynomial) hard problem and have used it effectively for mapping potential protein interactions. This computational approach can be used for studying organisms at systems level by integrating high throughput differential expression data and information on regulatory networks. This method can also be used to infer functional linkages and constructing genome-wide interactomes.
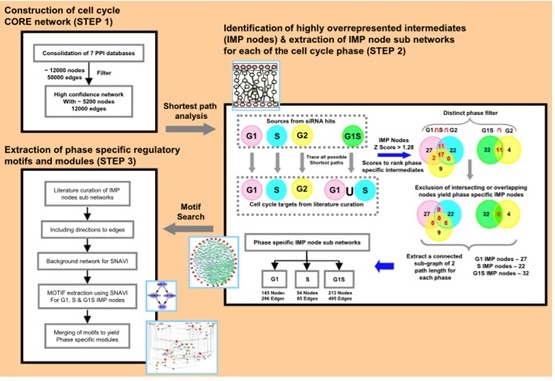 Figure 1: A systems approach for analysis of cell cycle regulation and the identification of phase-specific regulatory modules.
Figure 1: A systems approach for analysis of cell cycle regulation and the identification of phase-specific regulatory modules.
Some of the applications of this algorithm include mapping dynamics of protein-protein interaction namely signalling between protein kinases and transcription factors, identifying substrate for kinases, predicting transcription factors responsible for gene expression and identifying potential active sites of proteins. A web-based server PAR-3D has also been created for predicting the active site of proteins.
Reference:
- Understanding Communication Signals during Mycobacterial Latency through Predicted Genome-Wide Protein Interactions and Boolean Modeling, PLoS One. 2012;7(3):e33893 (Article).
- Delineation of key regulatory elements identifies points of vulnerability in the mitogen-activated signaling network, Genome Res. 2011 21: 2067-2081 (Article).
- Exploiting 3D structural templates for detection of metal-binding sites in protein structures, Proteins 2008;70:1206–1218 (Article).
- PAR-3D: a server to predict protein active site residues, Nucleic Acids Research, 2007, Vol. 35, Web Server issue W503–W505 (Article).
Technology Readiness: TRL B
Technology Status: Proprietary Know-how
Technology Availability: Know-how available for co-development.